 |
Chromosom 2 des Menschen
Ergebnis einer Fusion?
von Peer Terborg
Studium Integrale Journal
24. Jahrgang / Heft 1 - Mai 2017
Seite 12 - 21
|
 |
Zusammenfassung: Das menschliche Chromosom 2 scheint durch eine Verschmelzung zweier Chromosomen entstanden zu sein, die bei Menschenaffen getrennt vorkommen, und einige Indizien stützen dieses Szenario. Daher wird Chromosom 2 als starkes Argument für eine Abstammung des Menschen von affenartigen Vorfahren verwendet. Doch andere Befunde stellen diese Deutung infrage, insbesondere ein mehrfach funktionales Gen, das die Fusionsstelle enthält. Doch selbst wenn eine Fusion in der Vergangenheit stattgefunden hätte, wäre es kein Beweis für eine gemeinsame Abstammung.
|
|
|
Einleitung
Das Genom (Karyogramm*) des Menschen besteht aus 46 Chromosomen; das der heutigen großen Menschaffen (Schimpanse, Gorilla und Orang-Utan) aus 48. Die Tatsache, dass dem Menschen zwei Chromosomen fehlen, motivierte zur Formulierung einer Hypothese, dass in der Vergangenheit eine Chromosomenfusion erfolgt sein müsse. Damit würde die Plausibilität der mutmaßlichen gemeinsamen Abstammung dieser Organismen vergrößert. Als man vor etwa 50 Jahren die Anfärbung von Metaphase-Chromosomen so weit verbessern konnte, dass 320 bis 500 Banden pro haploidem Chromosomensatz identifizierbar wurden, wendete man diese neue Technik auch sehr bald an, um die Ähnlichkeit der Chromosomen und damit auch der vermuteten Verwandtschaftsverhältnisse zwischen Mensch und Menschenaffen zu bestimmen. Die vorgefundene generelle Homologie der Chromosomenbanden bei Mensch, Schimpanse, Gorilla und Orang-Utan stärkte die Hypothese, dass diese vier Spezies auf einen gemeinsamen Vorfahren zurückgeführt werden können. Eine vergleichbare hochauflösende Chromosomenfärbung legte 1982 nahe, dass 18 der 23 Chromosomen-Paare des modernen Menschen praktisch identisch mit denen der Menschenaffen sind, während die restlichen Chromosomenpaare Variationen des Heterochromatins aufweisen, die den unterschiedlichen Spezies die jeweils charakteristischen Eigenschaften verleihen. Im Vergleich mit dem Schimpansen wurden in neun der menschlichen Chromosomen (Chr1, -4, -5, -9, -12, -15, -16, -17 und -18) Inversionen* gefunden. Der prominenteste Unterschied, den diese Studien aufdeckten, war aber, dass alle Menschaffen 48 Chromosomen haben, während Menschen nur 46 besitzen. Die Chromosomenfärbungen identifizierten das menschliche Chr2 als ein Fusionsprodukt von zwei kleinen, in Menschaffen getrennt vorkommenden Chromosomen (Chr2A und Chr2B oder: 12 und 13) (Yunish & Prakash 1982). Obwohl eine Fusion der Telomere* der Chromosomen 2A und 2B die Reduktion von 24 Paaren von Chromosomen der großen Affen auf 23 beim modernen Menschen erklären würde, weisen Yunish & Prakash darauf hin, dass man die Fusion nur in Menschen vorfindet und sie daher nach der Abspaltung von Mensch und Schimpanse stattgefunden haben muss. Die Fusion an sich demonstriert die Plastizität des Genoms, beweist aber nicht eine Abstammung des Menschen von affenartigen Vorfahren.
 |
| Oft wird behauptet, genetische Daten würden eine Abstammung des Menschen von affenartigen Vorfahren beweisen. Die Datenlage ist viel zu kompliziert, als dass diese einfache Schlussfolgerung gezogen werden kann. (fotolia.com) |
|
|
Das menschliche Chromosom 2 (Chr2) wird typischerweise als Ergebnis eines alten Fusionsereignisses interpretiert. Zwei kleinere Chromosomen, die bei Menschenaffen getrennt vorkommen, sollen sich im Verlauf der Entwicklungsgeschichte vereinigt haben. Als Beleg werden dafür vor allem ein ähnliches Bandenmuster zwischen menschlichem Chr2 und den Schimpansen-Chromosomen Chr2A und Chr2B angeführt sowie ein Sequenzabschnitt, der als ein deaktiviertes Zentromer auf dem humanen Chr2 aufgefasst wird. Das angeblich deaktivierte Zentromer ist durch eine Reihe von 171-bp-Repeats* charakterisiert, die als Alpha-Satellit-DNA* bezeichnet wird. Außerdem enthält es eine Region mit vielen Telomer*-Wiederholungen (TTAGGG), die zunächst in einer bevorzugten Richtung laufen, gefolgt von spiegelbildlichen Kopien in die andere Richtung. Dies würde man genau so von einer Kopf-Kopf-Fusion erwarten. Die Chr2-Fusion ist eines der anschaulichsten Argumente für eine vermutete Abstammung des Menschen von affenähnlichen Vorfahren, wobei oft betont wird, dass sie tatsächlich die gemeinsame Abstammung von Menschen und Menschaffen beweise. Gemäß der aktuellen Genom-Karte und den ENCODE-Daten, die mit dem UCSC-Genome-Browser* abrufbar sind, befindet sich die sogenannte Fusionsstelle allerdings innerhalb des ersten Introns* des DDX11L2-Gens auf dem menschlichen Chr2. Dieses Gen ist wohl funktional, denn die mutmaßliche Fusionsstelle weist deutliche epigenetische und biochemische Aktivität auf und es befinden sich in der Fusionsstelle mehrere funktionelle Bindungsmotive für Transkriptionsfaktoren*. Die Bindungsstellen erlauben es, im Zusammenhang mit Polymerase-II-Komplexen, drei alternative DDX11L2-Gen-Transkripte hervorzubringen. Der Befund eines funktionierenden Gens stellt ein Fusionsereignis in Frage. Die Argumente, die für bzw. gegen eine mutmaßliche Chr2-Fusion sprechen, werden kritisch diskutiert und bewertet.
|
|
|
Hinweise auf eine Fusion: Telomer und Zentromer
Telomere sind Regionen am Ende der Chromosomen und enthalten Tausende von Wiederholungen der DNA-Sequenz „TTAGGG“. Sie binden an Proteine, die die Chromosomen vor Degeneration schützen. Wenn zwei Chromosomen an ihren Enden (Kopf-Kopf) fusionieren, so ergibt sich ein Chromosom mit zwei endständigen Telomeren sowie Telomer-Sequenzen im Bereich der Fusion des Chromosoms (Abb. 2). Des Weiteren resultiert aus einer Kopf-Kopf-Fusion zweier Chromosomen ein zusätzliches Zentromer* (Bereich der primären Einschnürungsstelle eines Metaphase-Chromosoms*). Mit den heute verfügbaren biotechnischen Methoden sind die beiden angesprochenen Merkmale schnell und leicht zu erkennen.
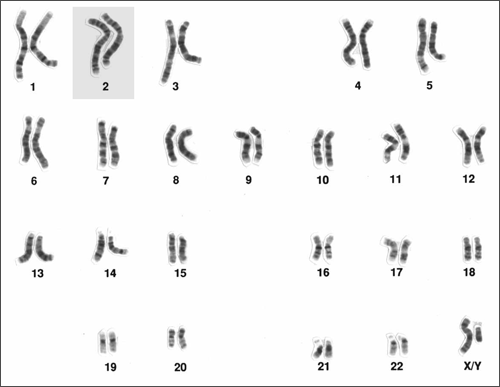 |
| Abb. 1: Die 46 Chromosomen des Menschen. Chromosom 2 ist unterlegt. (Wikimedia Commons) |
|
 |
| Die Fusion an sich demonstriert die Plastizität des Genoms, beweist aber nicht eine Abstammung des Menschen von affenartigen Vorfahren. |
 |
|
Chromosomale Aberrationen kommen beim Menschen in etwa 20 von 1000 Neugeborenen (Hamerton et al. 1976). Obwohl die meisten Träger von Fusionen gesund sind, besteht ein erhöhtes Risiko für Unfruchtbarkeit und spontane Abgänge aufgrund der Übertragung der unausgeglichenen Gameten (McKinlay Gardner 2012). Interessanterweise sind alle bekannten Fusionen in heute lebenden Tieren durch eine Sequenz gekennzeichnet, die als Satelliten-DNA (SatDNA) bezeichnet wird (Chaves et al. 2003; Tsipouri et al. 2008; Adega et al. 2009). Alle diese Fusionen sind das Ergebnis eines der beiden folgenden Szenarien: 1) SatDNA-SatDNA oder 2) SatDNA-telomereDNA (Tomkins 2013). Die Fusionssequenz, die wir im menschlichen Genom vorfinden, ist aber eine abnorme Telomer-Telomer-Fusion (TTF), die nur einmal in der wissenschaftlichen Literatur beschrieben worden ist – bei einem Fall von Darmkrebs (Tanaka et al. 2014). Beim Menschen sind verschiedene Fusionsereignisse bekannt, darunter Robertson‘sche Translokationen (Song et al. 2016). Sie haben zwar Auswirkungen auf die Fruchtbarkeit, verursachen aber keine Änderung im Phänotyp (Erscheinungsbild). Ebenso gibt es keinen Grund zur Annahme, dass eine Telomer-Fusion eine Affenart in einen Menschen umwandelt.
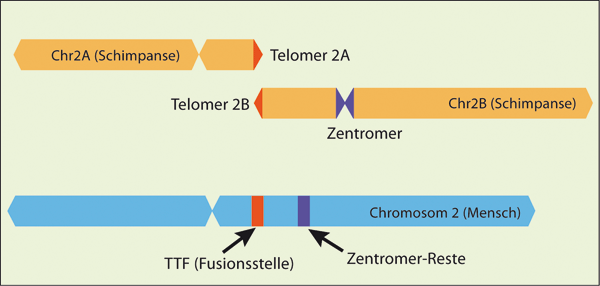 |
| Abb. 2: Darstellung der TTF, in der zwei Ur-Chromosomen (als Chr2A und Chr2B in Schimpansen-Genom) zu einem menschlichen Chromosom 2 fusionieren. Die Positionen von Zentromer und TFF auf Chromosom 2 basieren auf den aktuellen Stellen, die man im UCSC Genome Browser* vorfindet. Man beachte das extrem geringe Maß an Positionskorrespondenz für die kryptische Zentromerstelle. Bei der mutmaßlichen Fusion verschwanden etwa 24 Millionen Basen – d. h. etwa 10% der DNA-Sequenzen gingen verloren. Alle Chromosomen sind maßstabsgetreu gezeigt. (Nach Yunis & Prakash 1982) |
|
Die fast völlige Abwesenheit von dokumentierten TTF in heute lebenden Säugetieren ist weitgehend darauf zurückzuführen, dass Telomere mit einer hochspezifischen Sequenz abschließen, der sogenannten Endkappe. Diese als Shelterin-Protein-Komplex bezeichnete Struktur schützt das Chromosom vor einer Fusion (Tomkins 2011b). Trotzdem gibt es Hinweise auf eine derartige ungewöhnliche abnorme TTF im genetischen Sequenzbereich, in welchem die Fusion der Chromosomen lokalisiert sein soll. Die genaue chromosomale Position der Fusion wird mit den Koordinaten 2q13-2q14.1 bezeichnet, d.h. auf den langen Arm (q) des 2. Chromosoms (Abb. 2). Die Idee einer TTF kam 1991 auf, als 800 Basen dieser Stelle geklont, sequenziert und die Ergebnisse mittels einer Fusion zweier Telomere (TTF) erklärt wurden (Ijdo et al. 1991).
Ein Zentromer ist die eingeschnürte Region, die Wespentaille eines Metaphase-Chromosoms*, welche die Schwesterchromatiden zusammenhält (Abb. 2). Pro Chromosom gibt es in der Regel immer nur eines davon, weil das Zentromer als Anheftungsstelle der Spindelfasern dient, mit dessen Hilfe die duplizierten Chromosomen bei der Zellteilung auseinandergezogen werden. Falls zwei Chromosomen Kopf-Kopf fusionieren, erwartet man ein Fusionsprodukt mit zwei Zentromeren. Das menschliche Chr2 weist aber nur ein einziges Zentromer auf, was bedeutet, dass im Falle einer vorausgegangenen Fusion Relikte eines zweiten Zentromers im entsprechenden Genombereich vorhanden sein sollten. Diese erwartet man ungefähr an der betreffenden Stelle des Schimpansen-Chromosoms Chr2b (Abb. 2). Yunis und Prakash erforschten die mutmaßliche Fusionsregion im Jahr 1982 mit einer besonderen Technik, der Fluoreszenz-in-situ-Hybridisierung (abgekürzt: FISH), und fanden tatsächlich Hinweise auf ein zweites Zentromer auf Chr2 (Yunis & Prakash 1982). Das Vorkommen Zentromer-ähnlicher Sequenzen im entsprechenden Bereich des Chr2 wurde 1992 bestätigt (Ijdo et al. 1992). In der Wissenschaftswelt war es danach lange Zeit um dieses Thema ruhig, während die bis dahin vorliegenden Befunde vor allem in Internetforen als Beweise für einen gemeinsamen Vorfahren von Schimpanse und Mensch diskutiert wurden. Erst 2005, nachdem das menschliche Genom sowie Chr2A und Chr2B vom Schimpansen vollständig sequenziert waren, wurde dieses Thema in wissenschaftlichen Publikationen wieder intensiver diskutiert.
|
|
Neuere Daten
Im Jahr 2002 wurden 614.000 Nukleotide des Fusionsbereichs vollständig sequenziert und die Ergebnisse wurden in zwei Studien veröffentlicht (Fan et al. 2002a; 2002b). Die Autoren berichten von alpha-Satelliten*-ähnlichen DNA-Sequenzen, die sich etwa an der richtigen Stelle auf dem langen Arm des menschlichen Chr2 befinden, wenn man von einer vorausgegangenen Fusion ausgeht. Beim Menschen wird die Zentromer-Einheit, eine deutlich erkennbare Sequenz von genau 171 Nucleotiden, als alpha-Satellit-DNA bezeichnet. Die auf Chr2 vorkommenden Sequenzen könnten also als Relikte eines Zentromers interpretiert werden, wenn die Sequenzen sich am richtigen Ort befinden würden. Diese Erwartung ist jedoch nur sehr eingeschränkt erfüllt: Zwischen der Fusionsstelle und den als Zentromer-Relikt identifizierten Alpha-Satelliten fehlen etwa 24 Millionen Nukleotide im Vergleich mit Chr2A und Chr2B des Schimpansen. Zudem befindet sich im mutmaßlichen Fusionsbereich des menschlichen Chr2 eine einzigartige Sequenz von 150.000 Nukleotiden, die man nirgendwo im Schimpansen-Genom vorfindet. Solche Diskrepanzen kann man als Indels* bezeichnen, eine Folge von Mutationen, durch die DNA-Sequenzen hinzugefügt oder entfernt wurden. Diese Vorgänge sind allerdings wissenschaftlich nicht nachweisbar, da sie keine überprüfbaren Spuren hinterlassen. Überzeugender ist die Beobachtung von Dutzenden degenerierter 6-Basen-Paar-Motive, die Telomer-Wiederholungen zu sein scheinen und sich überwiegend in Vorwärtsrichtung (TTAGGG) oberhalb der Fusionsstelle und in der umgekehrten Richtung (CCCTAA) darunter befinden, genauso, wie es im Fusionsmodell erwartet wird (Fan et al. 2002a; 2002b). Die vermeintliche Fusionsstelle enthält aber keine SatDNA und beschränkt sich auf eine einzige Region von nur 798 Basen Länge, was sehr wenig ist im Vergleich zu dem, was man von einer Fusion Tausender Telomer-Wiederholungen erwarten würde (Tomkins und Bergman 2011).
Die beiden Studien aus dem Jahr 2002 brachten aber auch eine Reihe ziemlich schwerwiegender Probleme, die laut Genetiker Jeffrey Tomkins völlig im Widerspruch zum Fusionsmodell stehen (Tomkins 2013). Das erste Problem ist die fehlende Syntenie (Gemeinsamkeiten in der Reihenfolge von Genen oder Gensegmenten) der menschlichen Sequenz mit den Sequenzen beim Schimpansen. Die nur 798 BP (Basenpaare) lange mutmaßliche Fusionsstelle erwies sich als von vielen funktionalen Genen und mutmaßlichen Pseudogenen* umgeben, die keine Homologie mit ihren mutmaßlichen Herkunftsorten, den Enden der Schimpansen-Chromosomen 2A oder 2B, aufwiesen. Da Fan et al. keine Ähnlichkeit mit Genen des Schimpansen aus der Umgebung der vermuteten Fusionsstelle fanden, postulierten sie, dass nach dem Auftreten des Fusionsereignisses die Gene aus anderen Teilen des menschlichen Genoms übertragen wurden (Fan et al. 2002a).
|
| Es hätten zu viele evolutionäre Prozesse in relativ kurzer Zeit stattfinden müssen. |
|
|
Ein zweites Problem war, dass die mutmaßliche Fusionssequenz angesichts der abgeleiteten evolutionären Zeitskala sehr stark – zu stark – degeneriert war. Das heißt: Es hätten zu viele evolutionäre Prozesse in relativ kurzer Zeit stattfinden müssen. Die Forscher berichteten: „Nur 48 % der 127 Wiederholungen in RP11-395L14 (Bereich upstream* von der Fusionsstelle) und 46 % der 158 Wiederholungen in M73018 (Bereich downstream* von der Fusionsstelle) sind perfekte TTAGGG- oder TTGGGG-Einheiten“ (Fan et al. 2002a). Weitere Forscher berichteten: „(W)ährend der Bildung des menschlichen Chromosoms 2 wurde einer der beiden Zentromere inaktiviert (der mit dem des Schimpansen-Chr2B übereinstimmt), und die Struktur dieses Zentromers zerfiel rapide.“ Warum dieses Zentromer einer beschleunigten Degeneration ausgesetzt wurde, bleibt ungeklärt (Hillier et al. 2005). Zudem wurde festgestellt, dass die Region, die als Zentromer interpretiert wird, aus Sequenzen besteht, die auch auf zehn anderen menschlichen Chromosomen vorhanden sind, nämlich auf Chr1, -7, -9, -10, -13, -14, -15, -18, -21 und -22 (Hillier et al. 2005).
Drittens war die Entdeckung bemerkenswert, dass die Fusionsstelle innerhalb eines RNA-Helicase-artigen Pseudogens (heute als DDX11L2 bezeichnet) lag, eine Beobachtung, die von den Forschern weitgehend ignoriert wurde (Fan et al. 2002b). Seit 2005 wurde die Fusionsstelle des menschlichen Genoms mit verbesserten Annotationen* sowie einer erheblichen Anzahl von in Fachzeitschriften unveröffentlichten, jedoch öffentlich zugänglichen ENCODE*-Daten aktualisiert. Diese neuen Daten führten ebenfalls zu neuen Einsichten.
|
Alpha-Satelliten-DNA (Alpha-Satelliten-Sequenz): Bezeichnung für repetitive Einheiten (-> Repeats) im Genom, die in der Größenordnung von 60-200 Nukleotiden liegen. Sie sind für die Bildung wichtiger chromosomaler Strukturen wie z. B. der Zentromere zuständig. Annotation: In der Genetik und der Bioinformatik: Funktionelle Zuordnung, die sowohl aus experimentellen Befunden als auch aus einer bioinformatischen Voraussage stammen kann. Die Annotation einer DNA-Sequenz beschreibt die genaue Lage von Exons und Introns sowie die repetitiven und funktionellen DNA-Elemente in diesen Sequenzen. Denisova-Menschen: Ausgestorbene Frühmenschen aus dem Altai-Gebirge im südlichen Sibirien. Downstream: Unterhalb eines bestimmten Gens (oder einer bestimmten DNA-Sequenz). Exon: Der Teil eines Gens, der genetische Information enthält und in mRNA abgeschrieben wird und nach dem Spleißen erhalten bleibt. Exons tragen die biologische Funktion eines Proteins oder einer nichtcodierenden RNA. Angrenzende Exons werden durch Introns voneinander getrennt. exprimieren: Ablesen und Nutzen von Genen. Indel (Plural: Indels): Sammelbegriff für Insertion (Einfügung) oder Deletion (Verlust) von Basen in der DNA. Intron: Abschnitt der DNA innerhalb eines Gens, der benachbarte -> Exons trennt. Introns werden -> transkribiert (in prä-mRNA abgeschrieben), aber aus dieser herausgespleißt (ausgeschnitten), bevor sie in Proteine oder ncRNAs übersetzt wird. Inversion: Drehung eines chromosomalen Abschnitts um 180°. Karyogramm: Die geordneten Chromosomen eines Chromosomenpräparats, wie man sie während Zellteilungen in den Zellen vorfindet. Metaphase-Chromosom: Kondensiertes Chromosom, das wie ein X-förmiges Gebilde lichtmikroskopisch erkennbar ist. MicroRNA-Bindungsstelle: Sequenz in einem DNA-Abschnitt, die bei -> Transkription in mRNA MicroRNA-Moleküle bindet und somit die Expression dieses mRNAs beeinflusst. (MicroRNA-mRNA-Bindung initiiert vorwiegend einen beschleunigten Abbau von mRNA). Repeats: Bezeichnung für Wiederholungen von identischen oder sehr ähnlichen Sequenzmotiven in DNA oder RNA. Seed-Sequenz („Samensequenz“): Konservierte Sequenz mit einem spezifischen Merkmal (z. B. TTAGGG-Motive in Telomeren). Syntenisch: Gemeinsamkeiten in der Reihenfolge von DNA-Sequenzen auf verschiedenen chromosomalen Abschnitten beim Vergleich zwischen den Genomen zweier verschiedener Organismen. Telomer: Region am Ende eines Chromosoms, besteht aus Tausenden von Wiederholungen der DNA-Sequenz TTAGGG. Bindet Proteine, die die Chromosomen vor Degeneration schützen. Translation: Die „Übersetzung“ von mRNA in ein Protein. Translationsstelle: Sequenz in einem DNA-Abschnitt, die bei -> Transkription in mRNA für die Andockung des Translationskomplexes zuständig ist, damit die Anfertigung von Proteinen von dem richtigen AUG (Startcodon) des mRNA-Moleküls initiiert werden kann. Transkription: Das Abschreiben von DNA in mRNA. Transkriptionsfaktor: Komplex von Proteinen (und manchmal auch RNA), der das Abschreiben (Transkription) eines Gens initiiert, indem er an DNA bindet und einen Ansatzpunkt für die RNA-Polymerase bildet. Transkriptionsfaktor-Bindungsstelle: Sequenz in einem DNA-Abschnitt (oder in einem Gen) für die Andockung eines Transkriptionsfaktors, damit die Transkription vorbereitet oder initiiert wird. UCSC-Genome-Browser: Interaktive Online-Suchmaschine der Universität von Kalifornien, Santa Cruz (UCSC), USA; bietet Zugang zu Genomsequenzdaten aus einer Vielzahl von Modellorganismen einschließlich des Menschen. Upstream: Oberhalb eines bestimmten Gens oder einer bestimmten DNA-Sequenz. Zentromer: Bereich der primären Einschnürungsstelle (die sog. „Wespentaille“) eines -> Metaphase-Chromosoms.
|
|
|
|
|
Next Generation
 |
Abb. 3: Schematisierte grafische Darstellung der Bänderung (Idiogramm) des menschlichen Chromosoms 2. (National Library of Medicine)
|
|
Durch technische Entwicklungen im Bereich der DNA-Sequenzierung (Next Generation DNA-Sequenzierung) sowie leistungsfähigere Methoden der Bioinformatik konnte die Qualität von Genomanalysen extrem gesteigert werden. Tomkins und Bergman nutzten die entsprechenden Werkzeuge und stellten fest, dass eine Telomer-Signatur außerhalb des 798 BP langen Fusionsbereichs fast vollständig fehlt und TTAGGG/TTGGGG-Sequenzen nur vereinzelt angetroffen werden. Nach Tomkins sind menschliche Telomere typischerweise 5.000 bis 15.000 Basen lang, und ein TTF-Ereignis (s. o.) sollte eine Signatur von Tausenden von Basen hinterlassen haben (Tomkins & Bergman 2011). Die vermissten Sequenzabschnitte könnten aufgrund von Verlust-Mutationen fehlen, da diese kaum irgendwelchen Regeln folgen. Der Verlust genetischen Materials ist ein empirisch bestätigter evolutiver Mechanismus, und solange ein genetischer Verlust die Reproduktionsfähigkeit eines Organismus nicht beeinträchtigt, ist eine solche Erklärung möglich. Ferner bestätigten Tomkins und Bergman, dass innerhalb der 798-Basen-Sequenz der Nachweis für ein Fusionsereignis, wie oben diskutiert, stark eingeschränkt ist. Ein wichtiges Merkmal ist die Tatsache, dass die Telomer-Motive in der Fusionsstelle weitgehend monomer sind und nicht als kontinuierliche Wiederholungen gefunden werden. Von den 10 intakten TTAGGG-Motiven (Telomer-Repeats*) auf der linken Seite der 798-Basen-Fusionsstelle konnten nur zwei Tandem-Vorkommen nachgewiesen werden. Von den 43 intakten CCCTAA-Motiven (inverse Telomer-Repeats*) auf der rechten Seite der 798-Basen-Fusionsstelle werden zwölf Tandem-Motive gefunden. Als die 798-Basen-Fusionsstelle mit dem BLASTN-Algorithmus gegen das menschliche Genom durchsucht wurde, traten 85 Übereinstimmungen auf. Mit anderen Worten, es existieren 85 Fusions-ähnliche Sequenzen im Genom des Menschen. Viele dieser Motive sind nicht telomerisch lokalisiert und liegen innerhalb der Chromosomen, eine Beobachtung, die eine telomerische Herkunft fragwürdig macht. Diese internen Motive werden als interstitielle Telomersequenzen bezeichnet und sind bei Tieren und Menschen allgemein bekannt (Lin & Yan 2008). Eine alternative Erklärung für die an der vermeintlichen Chr2-Fusionsstelle vorhandenen Telomer-ähnlichen Merkmale ist daher, dass es sich dabei um eine Art von internen genomischen Motiven handelt. (Tomkins & Bergman 2011).
2012 wurde eine phylogenetische Studie veröffentlicht, in der die Fusionsstelle des Chr2 des Menschen mit den homologen Sequenzen des Schimpansen und des Gorillas verglichen wurde. Diese vergleichende genomische Analyse legte nahe, dass die Strukturen weitgehend unabhängig voneinander in den beiden Abstammungslinien entstanden sind, mit Ausnahme von wenigen sogenannten Seed-Sequenzen*, die sowohl in Menschen als auch in afrikanischen Affen vorkommen. Die Analyse, die die DNA-Sequenz und die genomische Organisation der chromosomalen Telomer-Endkappenregionen von Gorilla- und Schimpanse-Chromosomen charakterisierte, beschreibt große Mengen Schimpansen-spezifischer SatDNA an den Enden der Chromosomen 2A und 2B, die jedoch bei Menschen fehlten. Die Forscher schlussfolgerten: „Unsere Ergebnisse betonen das komplexe Zusammenspiel von duplizierten Sequenzen und chromosomalen Umlagerungen, das die zytogenetische Landschaft in kurzer evolutionärer Zeit schnell verändert“ (Ventura et al. 20121). Die Autoren vermuteten, dass die Schimpansen-SatDNA im Fusionsereignis verloren ging, doch die Telomer-Sequenz in einem von der Fusionsstelle weiter entfernten Bereich irgendwie verschont blieb. Trotz der vorgeschlagenen Verlust-Mutationen beim Fusionsereignis ist das von Ventura vorgestellte Modell nicht realistisch, wonach 24 Millionen DNA-Basen verloren gegangen sein sollen (Tomkins 2013). Auch hier gilt aber, obwohl es sich um einen Verlust von 24 Millionen Nukleotiden handelt, dass es keine Beschränkungen für Verlust-Mutationen gibt, solange sie die Reproduktion des Organismus nicht beeinträchtigen. Alles im allem haben die Fusions-Befürworter stärkere und schwächere Argumente, wobei die Telomer-Repeats am klarsten eine Fusion stützen, da sie anti-parallel verlaufen, genauso wie man erwarten würde, wenn in diesem Bereich eine TTF stattgefunden hat. Eine mutmaßliche TTF kann also tatsächlich in diesem Bereich verteidigt werden. Diese Schlussfolgerung bedeutet nicht automatisch eine Mensch-Schimpanse-Abstammung, da die Fusion eindeutig spezifisch für den Menschen ist und nicht in den heute lebenden Affen angetroffen wird. Auch Yunis & Prakash (1982) schließen, dass das Fusionsereignis nach der Aufspaltung dieser Organismen stattgefunden haben muss. Eine neue Studie zur Funktionalität der Fusionsstelle stellt die Fusion als Tatsache jedoch abermals in Frage, wie nachfolgend geschildert werden soll (Tomkins 2013).
|
|
Der DDX11L2-Gen-Locus
Seit der erstmaligen Sequenzierung des Fusionsbereichs auf dem Chromosom 2 (Fan et al. 2002a) wurde das Pseudogen, dessen DNA-Sequenz den Fusionsbereich beinhaltet, als eines der 18 Mitglieder der DDX11L-Familie von RNA-Helicase-Genen erkannt (Costa et al. 2009). Eine typische evolutionäre Erklärung zur Entstehung neuer Gene nimmt Bezug auf eine Duplikation bereits vorhandener ancestraler Gensequenzen.
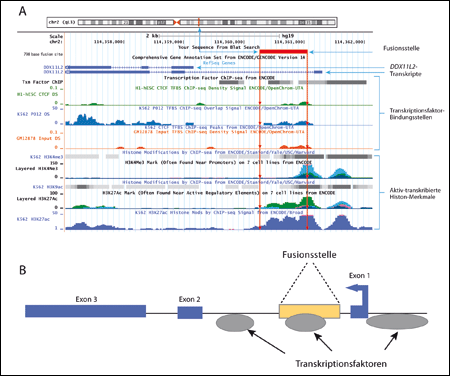 |
| Abb. 4: A UCSC-Genome-Browser-Daten, die ausgewählte Gen-Annotationen und ENCODE-bezogene Tracks für den DDX11L2-Gen-Locus zeigen, wobei die 798-Basen-Fusionsstelle innerhalb des Locus unter Verwendung von BLAT positioniert ist. Nach einem Analyse-Bild auf genome.ucsc.edu (Zugriff 23. Juli 2013). B Vereinfachte Darstellung der Fusionsstelle (rote Linie) innerhalb des DDX11L2-Gen-Locus. Der Pfeil im ersten Exon zeigt die Richtung der Transkription. Transkriptionsfaktor-Bindungstellen und Merkmale für aktiv-transkribiertes Chromatin befinden sich genau dort, wo sich die Fusionstelle befindet (zwischen den roten Pfeilen). (Nach Tomkins 2013) |
|
Costa und Mitarbeiter schlagen daher vor, dass alle Varianten der DDX11L-Gene beim Menschen aus Ahnensequenzen des Genoms von Affen entwickelt wurden. Diese Hypothese wurde vorgeschlagen, obwohl die Autoren nur wenige Stellen im Genom des Schimpansen und des Gorillas identifizierten, die als Vorläufersequenzen in Betracht kommen könnten. Beim Schimpansen befinden sich die betreffenden Stellen zudem nicht auf Chr2A oder Chr2B, sondern auf Chr12 und Chr20. Im Genom des Gorillas befinden sie sich auf den Chromosomen 3, 6, 7 und 20 (Costa et al. 2009). Die Tatsache, dass 18 variable Kopien des DDX11L-Gens beim Menschen existieren, aber nur zwei in Schimpansen und vier in Gorillas, stellt hohe Herausforderungen an die Flexibilität der hypothetischen evolutionären Prozesse der angenommenen Affe-Mensch-Evolution, besonders weil diese Sequenzen an völlig unterschiedlichen Positionen im jeweiligen Genom auftreten. Das bedeutet, dass im Blick auf das eben vorgestellte Phänomen ein erheblicher Aufwand an Hypothesen und Mutmaßungen betrieben werden muss, um eine gemeinsame Abstammung von Mensch und großen Menschenaffen plausibel zu machen. Die erforderlichen hypothetischen Mechanismen sind komplex und bisher liegen keine empirischen Befunde vor, die solche Vorgänge wahrscheinlich machen. Interessanterweise wurde in Costas Artikel, in dem die vermeintliche Entwicklungsgeschichte der DDX11L-Familie ausführlich beschrieben wurde, völlig ausgeblendet, dass das DDX11L2-Gen die bekannte Chromosom-2-Fusionssequenz enthält; eine seltsame und nur schwer verständliche Unterlassung, denn es ist genau die Fusionsstelle in diesem Gen, die als anschaulichster Beweis für die gemeinsame Abstammung mit den Menschaffen gewertet wird.
In seiner Studie aus dem Jahr 2003 analysierte Tomkins den DDX11L2-Gen-Locus mit Hilfe des UCSC-Genome-Browsers, das ist ein öffentliches Datensystem, das die neusten Gen-Annotationen und ENCODE-bezogene Tracks (d. h. Sequenzen) bereitstellt. Das System beinhaltet alle funktionellen Aspekte einer DNA-Sequenz wie z. B. Transkriptions*- und Translationsstellen*, Exons* und Introns*, miRNA-Bindungsstellen, epigenetische Marker usw. Die Datenbank belegt, dass der DDX11L2-Gen-Locus für drei Exons codiert und dass die Transkripte variabel und komplex sind (Tomkins 2013). Basierend auf der jüngsten Annotation des menschlichen Genoms (GRCh37/hg19; http://genome.ucsc.edu) zeigt Tomkins, dass die Fusionsstelle klar in dem ersten Intron des DDX11L2-Gens auf dem menschlichen Chromosom 2 enthalten ist (Abb. 4A, B). Das DDX11L2-Gen besteht aus drei Exons und wird von der Telomer-zu-Zentromer-Richtung auf dem Minus-Strang transkribiert (Abb. 4A, B). Somit wird die sogenannte Fusionssequenz im reversen Komplement als Teil eines funktionalen Gens tatsächlich abgelesen, allerdings nicht in der Vorwärtsstrang-Orientierung, wie sie typischerweise durch die sogenannte Fusions-Signatursequenz dargestellt ist – also 5‘->3‘ statt 3‘->5‘ (Tomkins 2013). Wenn die Fusionsstelle tatsächlich ein DNA-Bindungsstellenmotiv repräsentiert und der Schlüssel zur Funktion und Expression des DDX11L2-Gens ist, wie Tomkins vermutet, dann wäre es wichtig zu wissen, welche Transkripte produziert werden und wie eine zweite Promotorstelle im ersten Intron diesen Prozess beeinflusst. Der UCSC-Genome-Browser zeigt zwei Konsensus-Transkripte für das DDX11L2-Gen, die beide drei Exons enthalten, aber nur eines enthält das erste Exon direkt 5‘ (vor) dem Intron, das die Fusionssequenz enthält.2
Zusätzlich enthält die Fusionsstelle Sequenzen dreier unterschiedlicher funktionaler DNA-Bindungsdomänen, an denen Transkriptionsfaktoren* (CTCF, cMyc und Po12) binden, wie Chromatin-Immunpräzipitation-DNA-Sequenzierung (ChIP-seq)-Analysen belegen. Transkriptionsfaktoren sind Proteine, die an regulatorische Stellen in Genen oder in der näheren Umgebung bestimmter Gene binden, um deren Aktivität zu steuern, und funktionieren wie Ein-/Aus-Schalter. Es gibt tatsächlich drei Transkriptionsfaktor-Bindungssequenzen* im DDX11L2-Gen, wobei die beiden stärksten Bindungsbereiche in der Fusionsstelle auftreten und auch direkt 5‘ und proximal zum ersten Exon in der Promotorregion des Gens (Tomkins 2013). Diese beiden Transkriptionsfaktor-Bindungssequenzen stimmen mit spezifischen epigenetischen Markern überein, die mit der Transkriptionsaktivität assoziiert sind (Abb. 4A, B). Von besonderer Bedeutung ist das Vorhandensein spezifischer Modifikationen der Histone (Acetylierung von H3K27ac und H3K9ac und Methylierung von H3K4Me1, H3K4Me3), die an der Fusionsstelle und dem Genpromotorbereich identifiziert wurden und sich transkriptionell decken mit den Bereichen der Transkriptionsfaktorbindung. Typischerweise verbindet man die H3K27ac-Histon-Acetylierungen mit aktiven Genpromotoren (Dunham et al. 2012; Harmston & Lenhard 2013) und mit aktiven Enhancer-Elementen in weitreichenden Chromatin-Wechselwirkungen (Creyghton et al. 2010; Zentner et al. 2011). In Kombination mit den Transkriptionsfaktor-Bindungsstellen* markieren die Histon-Modifikationen die Fusionsstelle demzufolge als eine transkriptionell aktive Stelle. Mit anderen Worten, das DDX11L2-Gen wird häufig in zwei unterschiedliche RNA-Moleküle transkribiert (übersetzt).
|
|
Vernetzungen
Die von Tomkins durchgeführten Analysen kann man selber jederzeit nachvollziehen.3 Sie bestätigen, dass sich die Fusionsstelle innerhalb des DDX11L2-Gens auf Chr2 befindet. Dieses Gen enthält drei funktionale Transkriptionsfaktor-Bindungsstellen*, von denen sich eine in der mutmaßlichen Fusionsstelle befindet. Tomkins zeigte, dass die Fusionsstelle ein aktiver Promotor innerhalb des ersten Introns des DDX11L2-Gens ist. Dieser bindet zahlreiche Transkriptionsfaktoren und enthält mehrere aktive Polymerase-II-Transkriptions-Bindungsstellen*. Das DDX11L2-Gen wird aktiv abgelesen und produziert mehrere alternative RNA-Varianten, die aus zwei oder drei Exons bestehen. Es handelt sich also kaum um ein Helicase-artiges Pseudogen, eher betrifft es hier ein long non-coding (lnc)RNA-Gen, das exprimiert* und reguliert wird. Man muss sich immer klarmachen, dass Gene nie einzeln, isoliert funktionieren, sondern immer Teil eines biologischen Programms sind und in ein holistisches (ganzheitliches) Netzwerk eingebunden sind.
Zudem werden allgemein immer häufiger Funktionen für Pseudogene beschrieben, was daran erkannt werden kann, dass sie Transkripte produzieren, sogenannte lncRNAs*, die an der Regulation ihrer Protein-codierenden Homologa oder anderer Proteine im Netzwerk beteiligt sind. Es ist dabei von Bedeutung, dass die Proteine und ihre Regulatoren simultan und synchron exprimiert werden. Daher analysierte Tomkins Expressionsmuster für das Gen, das das DDX11-Protein codiert, in der Datenbank von BioGPS.org. Sowohl DDX11L2 als auch sein Homologon, DDX11, wurden in denselben Gewebe- und/oder Organkategorien exprimiert (Tomkins 2013).
|
| Die Fusionsstelle liegt innerhalb eines möglicherweise wichtigen und funktionalen Gens – ein Zufallsprodukt? |
|
|
Diese Daten stehen nicht für sich alleine. Auch die biomedizinische Datenbank genevestigator.com berichtet eine signifikante Expression von DDX11L2 in 255 verschiedenen Zell- und Gewebetypen (Hruz et al. 2008). Untersuchungen mit Daten aus der COXPRESSdb-Datenbank (Obayashi et al. 2013) zeigen, dass das DDX11L2-Gen zusammen mit einer Vielzahl unterschiedlicher Blutzell- und Chromatin-Remodelling-Gene exprimiert wird, ebenfalls gekoppelt mit der Expression des proteincodierenden DDX11-Gens.4 Diese Koexpressionsdaten liefern mögliche Hinweise auf die regulatorische Rolle von DDX11L2-Transkripten in Verbindung mit DDX11-proteincodierenden Transkripten. Hinweise darauf, in welche Art von generellen zellulären Prozessen das DDX11L2-Pseudogen eingebunden sein könnte, zeigten sich durch seine enge Verknüpfung mit mehreren anderen Genen. Die enge Kopplung der Koexpressionsdaten für DDX11L2 belegen, dass das Gen mit drei verschiedenen Schlüsselgenen, TREML1, TUBB1 und BEND2, transkriptionell verknüpft ist (Tomkins 2013). TREML1 spezifiziert einen Zelloberflächenrezeptor, der in myeloiden Zellen, d. h. nicht-lymphozytischen Zellen des Immunsystems, und bei der Gerinnung exprimiert wird. TUBB1 ist ein Mitglied der Beta-Tubulin-Proteinfamilie, spezifisch für Thrombozyten und Megakaryozyten, d. h. für die Knochenmarkszellen, die für die Produktion von Blutplättchen verantwortlich sind. Und BEND2 ist ein proteincodierendes Gen, das mit Protein-DNA-Wechselwirkungen assoziiert und während der Chromatin-Restrukturierung und Transkription aktiv ist. Grundsätzlich hat das DDX11L2-Gen alle Merkmale eines lncRNA-Gens, das im Zusammenhang mit mehreren Genen exprimiert wird und in deren Regulation involviert ist. Darüber hinaus deutet eine Vielzahl von MicroRNA-Bindungsstellen* auf eine komplexe posttranskriptionelle Regulation der unterschiedlichen DDX11L2-Gentranskripte hin. Es ist äußerst unwahrscheinlich, dass eine willkürliche Chromosomen-Fusion komplexe Multi-Exons und alternativ gespleißte funktionelle Gene hervorbringen könnte.
|
|
Belege für eine Fusion
Die jüngste Studie, die die Fusionsstelle beleuchtet, stammt aus dem Jahr 2016 (Miga 2016). Sie zeigt, dass sowohl der Neandertaler als auch der Denisova-Mensch* die 171bp Alpha-SatDNA-Wiederholungen aufweisen, die man auch in modernen Menschen findet. Die Sequenzen sind sogar sehr ähnlich, aber alle sind sehr verschieden von denjenigen in Schimpansen. Zudem bestätigte diese Studie, dass die Gensequenzen sich (nach dem evolutionären Modell) sehr schnell über kurze evolutionäre Zeit umgestaltet haben müssen. Die verschiedenen Arten haben auf dieser Sequenzebene keine Homologie: „... die vorherrschende SatDNA-Sequenz (D2Z1), die mit dem aktiven Zentromer auf dem menschlichen Chromosom 2 assoziiert ist, ist unähnlich dem Zentromer auf dem syntenischen* Chromosom 2A in Schimpansen ...“ (Miga 20165). Dem Anschein nach existieren sowohl Spezies-spezifische 171bp Alpha-SatDNA-Wiederholungen als auch Chromosom-spezifische Wiederholungen. Dementsprechend würde man erwarten, dass das rudimentäre Zentromer auf dem menschlichen Chr2 ganz anders – d. h. Spezies-spezifisch – ist als das funktionelle Zentromer auf dem Schimpansen-Chr2B. Das würde aber auch bedeuten, dass die Daten nicht als Belege für gemeinsame Abstammung gewertet werden können. Im darwinistischen Modell sollte das rudimentäre Zentromer degenerieren, aber seit der hypothetischen Mensch-Schimpanse-Aufspaltung sollte sich das Schimpansen-Chr2B-Zentromer auch verändert haben. Wenn die beiden Sequenzen heute verschieden sind, könnte dies als Indiz sowohl für gemeinsame Abstammung als auch für gemeinsames Design gewertet werden.
Die Autorin liefert jedoch zusätzliche Daten, um das Fusionsmodell zu unterstützen. Zwei Gene, die am Ende des Schimpansen-Chr2A und -2B gefunden wurden, flankieren das rudimentäre Zentromer beim Menschen in der erwarteten Entfernung und in der erwarteten Orientierung. Außerdem entsprechen Alpha-Sat-Monomere oberhalb und unterhalb des Zentromers denen des Schimpansen-Chr2A und -2B und es gibt zwei genomische Elemente (eine LINE und eine SVA), die auf Chr2A bzw. Chr2B gefunden wurden und das rudimentäre Zentromer des Menschen flankieren. Alles im allem zeigt Miga eine überzeugende Beweisführung für eine TTF.
|
|
Die Biologie braucht eine neue Theorie
Zieht man die bekannten molekularbiologischen und genetischen Daten in Betracht sowie die hier vorgestellten Argumente, dann wird deutlich, dass beim Thema Fusion des humanen Chr2 nach wie vor eine kontroverse Diskussion geführt wird. Einerseits gibt es sehr deutliche Hinweise auf eine TTF, andererseits gibt es klare Belege für die Existenz eines funktionalen Gens, das die Fusionsstelle beinhaltet, was gegen eine Fusion spricht.
|
| Der genetische Unterschied von Menschenaffen und Menschen betrifft viele Tausende von Genen. |
|
|
Entscheidend ist, wie man diese Daten insgesamt deutet. Geht man von dem (Neo-)Darwinistischen Paradigma aus, in dem zufällige genetische Abänderungen und eine gemeinsame Abstammung aller Organismen wesentlich sind, dann wird man eine Fusion des humanen Chr2 als Beweis für eine gemeinsame Abstammung mit den Menschaffen deuten. Dabei müssen aber die Daten zur Funktionalität des Gens im Fusionsbereich weitgehend ignoriert oder umgedeutet werden. Tomkins‘ Befunde wurden im Artikel von Karen Miga überhaupt nicht aufgenommen und diskutiert (Miga 2016). Die Tatsache, dass die Fusionsstelle innerhalb eines möglicherweise wichtigen und funktionalen Gens liegt, ist kaum mit der Hypothese vereinbar, dass sie das Zufallsprodukt einer genetischen Abnormität ist. Die enge Integration eines rezenten Mutationsereignisses in das Genom ist nahezu unmöglich in einem evolutionären Vorgang, in dem nur Anhäufungen von zufälligen Mutationen erlaubt sind. Eine zweite Studie aus 2016 geht davon aus, dass die Fusion ein einmaliges Ereignis war und dass der Verlust von Genen, die in die repetitiven subtelomerischen Sequenzen eingebettet waren, eine wichtige evolutionäre Rolle gespielt hat, indem er z. B. höhere kognitive Funktionen, aufrechte Körperhaltung, Ausdauerlauf oder verbesserte Thermoregulation durch eine verbesserte Transpirationsfähigkeit in verschiedenen Unterarten und Populationen von Homininen initiierte (Stankiewicz 2016). Mit anderen Worten: Die Höherentwicklung des Menschen wurde durch Verlust von biologischer Information vorangetrieben – eine sehr unorthodoxe Sichtweise.
|
| Die Befunde könnten auch ein gemeinsames Design oder einen gemeinsamen biogenetischen Mechanismus widerspiegeln. |
|
|
Die derzeitigen Erkenntnisse dokumentieren, dass der genetische Unterschied von Menschenaffen und Menschen viele Tausende von Genen betrifft. Es gibt 643 Menschen-spezifische und 780 Schimpansen-spezifische proteincodierende Gene und dazu noch etwa 1000 microRNA-Gene, die man nur beim Menschen antrifft. Solche Befunde sind nur schwer verträglich mit einer gemeinsamen Abstammung dieser Arten. Zudem belegen vergleichende Genomstudien, in denen man etwa 2700 human accelerated regions (HARs) feststellte, dass genetische Veränderungen extrem schnell verlaufen sein müssten, wenn man von einer Abstammung von Menschen und heutigen Menschenaffen von gemeinsamen Vorfahren in den angenommenen Zeiträumen ausgeht. Daten, die als Hinweis auf eine gemeinsame Abstammung interpretiert werden, sollten demgemäß mit mehr kritischer Zurückhaltung betrachtet werden (wie z. B. bei Tomkins). Die Befunde könnten ja auch ein gemeinsames Design oder einen gemeinsamen biogenetischen Mechanismus widerspiegeln (Terborg 2010). Die Frage nach der Herkunft der jeweils spezifischen Information, die sich im Genom von Menschen und großen Menschenaffen widerspiegelt, wird in der aktuellen Diskussion der Biowissenschaften nicht beantwortet. Könnte es sein, dass etablierte darwinistische Denkmuster neue Hypothesen hemmen? Vermisst wird in der Biologie eine Informationstheorie, die den Ursprung der biologischen, in den Genomen niedergelegten Information sowie deren Modifikation und Konservierung behandelt. Die Biologie braucht die Einsicht, dass manche evolutionäre Prozesse programmiert (front-loaded) sein können. Demnach wäre die Information programmiert und bestimmte genetische Veränderungen erfolgen nicht nur zufällig und ungerichtet (Terborg 2008; 2009). Eine solche Theorie könnte vermutlich vieles erklären, was jetzt offen bleiben muss.
Dank
Besonderer Dank geht an Dr. Jeffrey G. Tomkins für fruchtbare Informationen, Hinweise und Abbildungen.
|
Anmerkungen und Originalzitate
1 Anti-Schöpfungs-Blogger behaupten, dass Tomkins falsch liegt, aber man kann seine Daten selbst validieren: http://genome.ucsc.edu/cgi-bin/hgGateway. Man setzt bei genome-euro.ucsc.edu die „human assembly“ auf „hg19“ (dies ist kritisch), gibt die Fusion-Site-Koordinaten ein: „chr2: 114,360,257-114,361,054“, und vergrößert sie durch Wahl von base mit der „ZOOM IN“-Funktion, um sicherzustellen, dass man die Nukleotid-Sequenz anschauen kann. Jetzt verkleinern, bis sichtbar wird, welche Gene und mRNAs in dieser Region annotiert sind. Hier findet man das DDX11L2-Gen; die Fusion ist genau in der Mitte des ersten Introns.
2 DDX11 ist in der COXPRESSdb-Datenbank auf Stelle 27 der Koexpressionsliste des DDX11L2-Gens zu finden. URL = http://coxpresdb.jp/
3 Ventura et al. (2012): „Our findings highlight the complex interplay between duplicated sequences and chromosomal rearrangements that rapidly alter the cytogenetic landscape in a short period of evolutionary time.“
4 In der NCBI-Nukleotid-Datenbank entsprechen diese DDX11L2-Gensequenzen den Zugängen NR_024005.2 und NR_024004.1. Die RNA-Transkripte haben eine Länge von 1.668 bzw. 2.188 Nukleotiden. Das längste Transkript von 2.158 Basen bildet die gesamte Länge des DDX11L2-Gens ab.
5 Miga (2016): „… the predominant satellite array (D2Z1) associated with the active centromere on human chromosome 2 is distinct from the HOR (i.e. high order repeats of a 171-bp alpha satellite DNA) array on the syntenic chimpanzee chromosome 2A centromere …“
|
|
|
Literatur
- Adega F, Guedes-Pinto H & Chaves R (2009)
- Satellite DNA in the karyotype evolution of domestic animals – clinical considerations. Cytogen. Genome Res. 126, 12-20.
- Chaves R, Adega F, Wienberg J, Guedes-Pinto H & Heslop-Harrison SJ (2003)
- Molecular cytogenetic analysis and centromeric satellite organization of a novel 8;11 translocation in sheep: A possible intermediate in biarmed chromosome evolution. Mammalian Genome 14, 706-710.
- Costa V, Casamassimi A, Roberto R, Gianfrancesco F, Matarazzo MR, D’Urso M, D’Esposito M, Rocchi M & Ciccodicola A (2009)
- DDX11L: A novel transcript family emerging from human subtelomeric regions. BMC Genomics 10:250.
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010)
- Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931-21936.
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. (2012)
- An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57-74.
- Fairbanks DJ (2010)
- Relics of Eden: The powerful evidence of evolution in human DNA. Amherst, New York: Prometheus Books.
- Fan Y, Linardopoulou E, Friedman C, Williams E & Trask BJ (2002a)
- Genomic structure and evolution of the ancestral chromosome fusion site in 2q13-2q14.1 and paralogous regions on other human chromosomes. Genome Res. 12, 1651-1662.
- Fan Y, Newman T, Linardopoulou E & Trask BJ (2002b)
- Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13-2q14.1 and paralogous regions. Genome Res. 12, 1663-1672.
- Hamerton JL, Canning N, Ray M & Smith S (1975)
- A cytogenetic survey of 14,069 newborn infants. I. Incidence of chromosome abnormalities. Clin. Genet. 8, 223-243.
- Harmston N & Lenhard B (2013)
- Chromatin and epigenetic features of long-range gene regulation. Nucleic Acids Res. 15, 7185-7199.
- Hillier LW, et al. (2005)
- Generation and annotation of the DNA sequences of humans chromosomes 2 and 4. Nature 434, 724-731.
- Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, Widmayer P, Gruissem W & Zimmermann P (2008)
- Genevestigator v3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008:420747, doi:10.1155/2008/420747.
- Ijdo JW, et al. (1991)
- Origin of human chromosome 2: an ancestral telomere-telomere fusion. Proc. Natl. Acad. Sci. USA 88, 9051-9055.
- Lin WL & Yan J (2008)
- Endings in the middle: Current knowledge of interstitial telomeric sequences. Mutation Res. 658, 95-110.
- McKinlay Gardner RJ, Sutherland GR & Shaffer LG (2012)
- Robertsonian translocations. In: Chromosome abnormalities and genetic counseling. New York: Oxford University Press, pp. 140-154.
- Melkikh AV & Khrennikov A (2017)
- Quantum-like model of partially directed evolution. Progr. Biophys. Mol. Biol., doi: 10.1016/j.pbiomolbio.2016.12.005.
- Miga KH (2016)
- Chromosome-specific centromere sequences provide an estimate of the ancestral chromosome 2 fusion event in hominin genomes. J. Hered. 108, 45-52, doi:10.1093/jhered/esw039.
- Obayashi T, Okamura Y, Ito S, Tadaka S, Motoike IN & Kinoshita K (2013)
- COXPRESdb: A database of comparative gene coexpression networks of eleven species for mammals. Nucleic Acids Res. 41 (Database issue), D1014-1420, doi: 10.1093/nar/gks1014.
- Song J, Li X, et al. (2016)
- A family with Robertsonian translocation: a potential mechanism of speciation in humans. Mol. Cytogen. 9:48, doi:10.1186/s13039-016-0255-7.
- Stankiewicz P (2016)
- One pedigree we all may have come from – did Adam and Eve have the chromosome 2 fusion? Mol. Cytogenet. 9:72.
- Tanaka H, Beam MJ & Caruana K (2014)
- The presence of telomere fusion in sporadic colon cancer independently of disease stage, TP53/KRAS mutation status, mean telomere length, and telomerase activity. Neoplasia 16, 814-823.
- Terborg P (2008a)
- Evidence for the design of life: Part 1. Genetic Redundancy. J. Creation 22,79-84.
- Terborg P (2008b)
- Evidence for the design of life: Part 2. Baranomes. J. Creation 22, 68-76.
- Terborg P (2009)
- Evidence for the design of life: Part 3. An introduction to variation-inducing genetic elements. J. Creation 23, 99-106.
- Terborg P (2010)
- An illusion of common descent. J. Creation 24, 122-127.
- Tomkins JG & Bergman J (2011)
- The chromosome 2 fusion model of human evolution. Part 2: Re-analysis of the genomic data. J. Creation 25,111-117.
- Tomkins JG (2013)
- Alleged human chromosome 2 “fusion site” encodes an active DNA binding domain inside a complex and highly expressed gene – negating fusion. Answers Res. J. 6, 367-375. https://assets.answersingenesis.org/doc/articles/pdf-versions/arj/v6/human-chromosome-fusion.pdf
- Tsipouri V, Schueler MG, Hu S, NIS Comparative Sequencing Program, Dutra A, Pak E, Riethman H, & Green ED (2008)
- Comparative sequence analyses reveal sites of ancestral chromosomal fusions in the Indian muntjac genome. Genome Biol. 9, R155, doi:10.1186/gb-2008-9-10-r155.
- Ventura M, Catacchio CR, Sajjadian S, Vives L, Sudmant PH, Marques-Bonet T, Graves TA, Wilson RK & Eichler EE (2012)
- The evolution of African great ape subtelomeric heterochromatin and the fusion of human chromosome 2. Genome Res. 22, 1036-1049.
- Yunis JJ & Prakash O (1982)
- The origin of man: a chromosomal pictorial legacy. Science 215, 1525-1530.
- Zentner GE, Tesar PJ & Scacheri PC (2011)
- Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 21, 1273-1283.
|
|
|  |
