 |
Die Suche nach der Nadel im Heuhaufen
von Matti Leisola
Studium Integrale Journal
21. Jahrgang / Heft 2 - Oktober 2014
Seite 99 - 103
|
 |
Zusammenfassung: Modelle zur Lebensentstehung müssen u. a. die Frage nach der Herkunft der Enzyme plausibel beantworten. Proteine, die durch ihre Struktur in der Lage sind, biochemische Reaktionen zu katalysieren, müssen früher oder später zur Verfügung stehen. Moderne Labormethoden ermöglichen Synthesen von Protein-Bibliotheken mit einer unüberschaubaren Vielfalt an Proteinen, die sich in ihrer Sequenz unterscheiden. Welche Hinweise liefern die bisherigen Erkenntnisse für eine mögliche Entstehung erster funktionsfähiger Proteine?
| |
|
Einführung
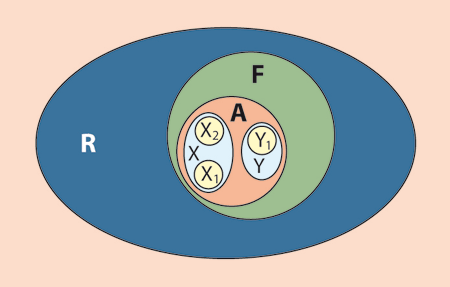 |
| Abb. 1: Vereinfachte und schematische Darstellung des Sequenzraumes R von Aminosäuresequenzen mit verschiedenen Fraktionen. Es gibt eine gewisse Anzahl von Sequenzen (Bereich F), die sich in eine bestimmte stabile Raumstruktur falten können. Eine stabile Faltung ist eine notwendige, aber noch keine hinreichende Bedingung für eine biologische Funktion. Die Fraktion mit einer biologischen Aktivität ist durch die Ellipse A symbolisiert. Dieser Bereich soll alle biologischen Aktivitäten umfassen, seien es Enzym-, Struktur- oder Regulationsfunktionen. Unter diesen gibt es Proteine mit bestimmten Funktionen, z. B. einer b-Lactamase-Funktion (Ellipse X). Es muss aber auch beachtet werden, dass eine bestimmte Funktion von unterschiedlichen Faltungstypen, die möglicherweise keinerlei Sequenzähnlichkeit zueinander haben, ausgeführt werden kann (z. B. X1 und X2). In Ellipse A befinden sich neben der Funktion X tausende von verschiedenen Funktionstypen, z. B. Funktion Y. |
|
Enzyme sind dadurch charakterisiert, dass sie komplexe molekulare Prozesse des Stoffwechsels mit hoher Spezifität und Effektivität katalysieren. Könnten Menschen Proteine auf vergleichbarem Niveau entwickeln, würde dies erstaunliche Möglichkeiten in der Biotechnologie eröffnen, z. B. Enzyme zur Synthese ganz spezieller Produkte oder zum gezielten Abbau von Problemstoffen. Als besondere Herausforderung erscheint bei Enzymen, dass trotz der Größe ihrer Struktur die eigentliche katalytische Aktivität auf einen kleinen Bereich der Gesamtstruktur begrenzt ist. Ein Protein durchschnittlicher Größe ist aus ca. 300 Aminosäuren aufgebaut, die in einer riesigen Zahl von Variationen (20300) kombiniert werden können. Diese enorme Vielfalt stellt den Sequenzraum dar (Abb. 1). Die Vorstellung, dass katalytische Aktivität unter zufällig angeordneten Polypeptiden (lange Kettenmoleküle aus Aminosäuren) ziemlich häufig auftreten könnte, geht zurück auf die Arbeit von Sidney Fox über Protein-Mikrosphären in den 1950er Jahren (Fox & Kaoru 1958). Wie ist diese Idee heute, fast 60 Jahre später, zu bewerten? Die bekanntesten neueren Arbeiten, die sich mit dieser Position auseinandersetzen, wurden im Labor des Nobelpreisträgers Jack Szostak seit den frühen 1990er Jahren durchgeführt. 60 Jahre experimentelle Forschung haben uns jedoch dem Ziel nicht näher gebracht, enzymatische Aktivität in Polypeptiden mit zufälliger Sequenz zu finden.
Allgemein stimmen Naturwissenschaftler darin überein, dass nur ein sehr kleiner Teil der möglichen Proteinstrukturen eine für entsprechende Funktionen notwendige dreidimensionale Struktur ausbildet (vgl. Abb. 1). Wie groß dieser Anteil ist, wurde in der Fachliteratur umfangreich diskutiert. Auf der Basis von bekannten Cytochrom C-Sequenzen bestimmte Hubert Yockey (1979) diesen Anteil mit 10-65 als äußerst gering. Reidhaar-Olson & Sauer (1990) errechneten einen Anteil von 10-63. Später leitete Douglas Axe (2004) von seinen Untersuchungen über Penicillin abbauende β-Lactamasen ab, dass die Wahrscheinlichkeit, ein funktionsfähiges Enzym unter zufälligen Sequenzen zu finden, ungefähr 10-77 - 10-53 beträgt. In kritischen Übersichtsbeiträgen kamen Axe (2010) und Kozulic (2011) zu dem Schluss, dass funktionale Proteine außerhalb der Reichweite von zufälligen Reaktionen liegen.
|
|
Zufallssequenzen, die an ATP binden
Die Arbeitsgruppe des Nobelpreisträgers Jack Szostak ist eine der wenigen Forschungsgruppen, die in experimentellen Studien versuchen, in zufällig erzeugten Sequenzen RNA- und Protein-Moleküle zu finden, die eine biologische Funktion aufweisen. Seit über 20 Jahren erstellt die Arbeitsgruppe Bibliotheken von bis zu 1013 Molekülen (Bartel & Szostak 1993). Durch Nutzung von sehr leistungsfähigen in vitro Selektionsmethoden, in welchen die Proteine mit der jeweils codierenden mRNA verknüpft werden (Roberts & Szostak 1997), durchlief eine Bibliothek von fast 1013 Proteinsequenzen mehrere Zyklen von zufälliger Veränderung (Mutagenese) und Selektion auf ATP-Affinität (Keefe & Szostak 2001). Schließlich erhielten die Autoren ein kurzes Protein, das eine signifikante ATP-Bindungsaktivität zeigte, obwohl es nur geringe strukturelle Stabilität aufwies. Aufgrund dieser Resultate kamen sie zu dem Schluss, dass funktionelle Sequenzen mit einer Wahrscheinlichkeit von 10-11 gefunden werden können, also unter 1011 Proteinen mit unterschiedlichen Sequenzen eine mit ATP-Bindungsfähigkeit. Diese Wahrscheinlichkeit ist verglichen mit den oben genannten hoch. Eine verkürzte Version dieses Proteins, von den Autoren als „artificial nucleotide-binding protein“, ANBP bezeichnet, war strukturell stabil genug, um es zu kristallisieren, und so konnten die Wissenschaftler den ersten Bericht über die Struktur eines Proteins aus einer Zufallsbibliothek vorstellen (Lo Surdo, Walsh & Sollazo 2004). Im Vergleich zu anderen ATP-bindenden Proteinen zeigt die Struktur von ANBP jedoch ein sehr eng begrenztes Potential als Enzym. Die Bedeutung dieser Veröffentlichung wurde in einem Übersichtsartikel über Protein-Engineering (Leisola & Turunen 2007) kurz diskutiert.
|
|
Information aufgrund von Zufall?
Als die eben genannte zufällig erzeugte ATP-bindende Aminosäuresequenz später in Zellen synthetisiert (exprimiert) wurde, entstanden jedoch Probleme, und die Zellen waren nicht lebensfähig (Stomel et al. 2009). Wir können daraus ableiten, dass Zellen mit dieser Sequenz keine Nachkommen hinterlassen hätten. Bei genauerer Analyse der Arbeiten von Szostaks Arbeitsgruppe zeigt sich auch, dass diese Experimente insofern nicht wirklich zufällig waren, als beträchtlicher Einsatz in Form von konzeptionellen Überlegungen und Information eingebracht wurde. In lebenden Systemen außerhalb des Labors wäre die Wahrscheinlichkeit für die Entstehung dieser ATP-bindenden Struktur viel kleiner als 10-11, da zufällige Mutationen in der DNA Stopp-Codons produziert hätten, an welchen die Proteinbiosynthese abgebrochen wäre und die dadurch verursachte Verschiebung des Leserahmens für die Nukleotid-Tripletts katastrophale Konsequenzen nach sich gezogen hätte. Die Experimente waren bewusst so konzipiert, dass solche Möglichkeiten verhindert werden. Demnach ist es wichtig, zwischen solchen Experimenten im Reagenzglas (in vitro) und Mutationen in natürlichen Systemen (in vivo) zu unterscheiden.
 |
| Experimente, in die ein beträchtlicher Einsatz in Form von konzeptionellen Überlegungen und Information eingebracht wird, bilden keine Zufallsprozesse ab. |
 |
|
Mutationsexperimente mit natürlichen Systemen zeigen, dass nur eine von 10 Milliarden aktiven Mutanten der Triosephosphatisomerase in zellulären Zusammenhängen ordentlich funktioniert (Silverman, Balakrishnan & Harbury 2001). Die Ursache dafür könnte darin liegen, dass sich selbst funktionsfähige Sequenzen in Zellen nicht richtig falten. Sie ballen sich leicht zusammen und bleiben so nicht in Lösung. Aus diesem Grund haben Szostak et al. nur kurze Aminosäureketten, Zufallssequenzen aus 80 Aminosäuren hergestellt. Lebende Zellen enthalten komplexe biomolekulare Maschinen, die als Chaperone bezeichnet werden und die große Proteine darin unterstützen, sich richtig zu falten. Darüber hinaus ist es keine Überraschung, dass zufällige Proteinstrukturen aufgrund der vielen chemisch funktionalen Seitenketten der Aminosäuren ATP binden. Proteine, die ATP und andere Nukleotide binden, sind in der Natur sehr verbreitet: es gibt mehr als 70.000 sogenannter Rossman-Domänen, die NAD(P) binden und fast 200.000 Nukleotidphosphat-Hydrolasen (Dessailly et al. 2009). Die Bindung von ATP ist biochemisch in Stoffwechselsystemen sehr häufig und stellt eigentlich kein repräsentatives Beispiel für Funktionalität von Proteinen dar.
Funktionsfähige Sequenzen, die den natürlichen Proteinen ähnlich sind, scheinen also in den Bibliotheken mit Zufallssequenzen sehr selten zu sein. Proteine, die Faltung zeigen, neigen zu Problemen mit der Löslichkeit (Doi et al. 2005). Selbst wenn man die N-terminale Hälfte des Kälteschock-Proteins CspA mit gleich langen Zufallssequenzen verknüpft (Riechmann & Winter 2000), erhält man nur sehr wenige gefaltete Proteine (eines von 107). Als Schlussfolgerung aus diesen Beobachtungen kann man sagen, dass die völlig zufällige Suche kaum ein geeigneter Weg zu sein scheint, um neue Proteine für den Einsatz in der Biotechnologie zu finden. Das hält uns jedoch nicht davon ab, mittels zufälliger Methoden die Umgebung von bekannten funktionalen Proteinstrukturen zu erforschen.
|
|
Zufallsmethoden in begrenztem Sequenzraum
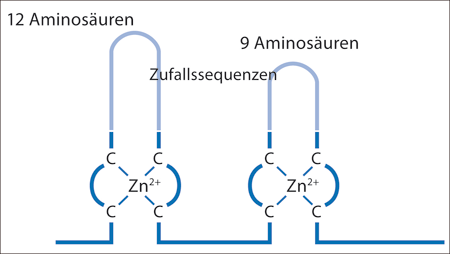 |
| Abb. 2: Zinkfingerdomänen stabilisieren zufällige Schleifen. (Nach Seelig & Szostak 2007) |
|
Einige eindrückliche praktische Erfolge wurden mit sogenannten gesteuerten Evolutionsmethoden erzielt (Leisola & Turunen 2006). Dabei ist der Ausdruck „gesteuerte Evolution“ ein Widerspruch in sich selbst, da eine gesteuerte Evolution nicht mehr zufällig ist. Der Ausgangspunkt für Experimente dieser Art ist ein funktionsfähiges, richtig gefaltetes Protein. Die Erforschung bezieht sich somit nur auf einen kleinen begrenzten Bereich. Darüber hinaus muss eine Abfolge der Mutationen existieren, die über richtig gefaltete, selektierbare Varianten zum gewünschten Punkt führt, sonst werden zufällige Mutationen die Information für enzymatische Aktivität rasch zerstören, wodurch der weitere Weg abgebrochen wird.
Experimente dieser Art haben Szostak et al. in ihren neueren Untersuchungen durchgeführt und dadurch etwas erzeugt, was sie als neue enzymatische Aktivität bezeichnen (Seelig & Szostak 2007; Chao et al. 2013). Sie behaupten, damit sei erstmals mittels Zufall eine neue Ligase-Aktivität erzeugt worden. Wie können wir diese Studie verstehen?
Als Ausgangspunkt für ihre Untersuchung wählten Seelig & Szostak die aus 460 Aminosäuren bestehende Sequenz des Retinoid-X-Rezeptors, RXRa, die in der Natur an DNA bindet. In zwei kleinen Schleifen wurden insgesamt 21 Aminosäuren zufällig ausgetauscht, wobei eine Bibliothek aus 1012 Zufallssequenzen (bezogen auf die 21 Aminosäuren) erzeugt wurde. Die Schleifen waren als Zinkfingerdomänen stabilisiert (Abb. 2). Damit ist, wenn überhaupt, nur ein kleiner Beitrag aus dem Bereich der Schleife selbst für die stabile Faltung erforderlich. Unter den erzeugten Zufallssequenzen zeigten einige Ligaseaktivität, d. h. aktivierte RNA-Moleküle können mit weiterer RNA verknüpft werden. Diese Aktivität wurde dann durch gerichtete Evolutionsmethoden weiter optimiert.
Die Autoren verglichen dann die erzielte Ligaseaktivität mit den spontan ablaufenden, durch Mg2+ katalysierten Verknüpfungsreaktionen und schlossen auf eine um mindestens 2x106 höhere Aktivität. Wie verhält sich dieses künstlich erzeugte Enzym aber im Vergleich zu natürlichen Ligasen? Eine Studie über die Kinetik einer Ligase wurde vor kurzem veröffentlicht (Gregory 2011). Für T4-DNA-Ligase, die an DNA-Doppelhelices Phosphorsäurediesterbindungen erzeugt, wird eine Katalysekonstante kkat von annähernd 1 s-1 und die Michaeliskonstante Km mit ca. 3 nM angegeben. Das von Seelig & Szostak beschriebene „Enzym“ hat eine kkat von ca. 1 h-1 (also ungefähr 3600 mal geringer). Die entsprechende Michaeliskonstante wird von den Autoren nicht angegeben. Sie schreiben aber, dass bei 10 mM Substrat noch keine Sättigung erreicht ist. Das bedeutet, dass Km mindestens so hoch, d. h. größer als 10 mM ist, was wiederum ein gut 3.000-fach schlechterer Wert als bei T4-Ligase ist. Der Wert für kkat /Km für dieses sogenannte „Enzym“ ist daher im Vergleich zur natürlichen T4-Ligase 10 Millionen mal schlechter. Hätten die Autoren ihre Befunde mit realen Enzymen verglichen, wäre deutlich geworden, dass sie nicht wirklich ein Enzym haben.
Darüber hinaus wurde bei dem genannten Experiment eine Vereinfachung für den mehrstufigen Prozess eingeführt (natürliche Ligasen müssen, bevor sie Phosphorsäurediester bilden können, mit ATP reagieren). Im Experiment wurde dieser Schritt durch den Einbau von energiereichem Triphosphat in das Substrat umgangen. Legt man die künstliche Struktur und die nichtnatürliche Reaktion, die katalysiert wird, zugrunde, wird deutlich, dass das erzeugte „Enzym“ in einer lebendigen Zelle nicht funktionieren würde. Das Experiment ist kein Beispiel für einen zufälligen evolutionären Prozess. Es wurde intelligent konzipiert und ausgeführt, außerdem wurden dafür Methoden und Bibliotheken genutzt, die seit über 20 Jahren entwickelt werden. Truman hat die Studien von Szostak et al. unter dem Aspekt der Information analysiert (Truman 2013).
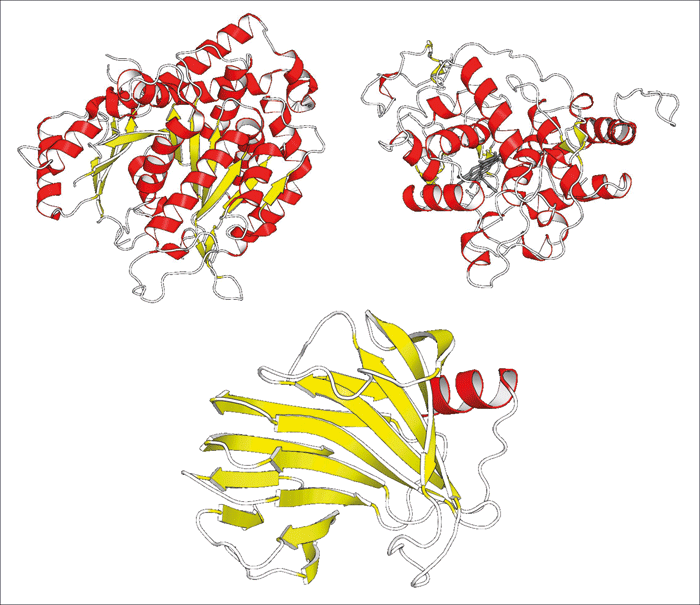 |
| Abb. 3: Drei Beispiele von holzabbauenden Enzymen, mit denen der Autor gearbeitet hat, in dreidimensionaler modellhafter Darstellung. Oben links: Cellubiohydrolase; rechts: Lignin-Peroxidase, unten: Xylase. |
|
|
|
Was wäre wenn?
Nehmen wir an, dass die beschriebene Abfolge von Mutationen sich in einer lebenden Zelle ereignet hätte. Wäre dies ein Beweis für zufällige Evolution? Seelig & Szostak erzeugten 4x1012 mutierte Proteine. Nach Analyse der aktiven Sequenzen schlossen die Autoren, dass 9 der 21 Aminosäuren für die beobachtete Funktion verantwortlich waren. Von diesen waren nur vier in allen aktiven Sequenzen vorhanden. Nehmen wir an, dass die Mutationshäufigkeit bei Escherichia coli unter künstlichen Bedingungen 10-7 pro Nukleotid und pro Zellteilung beträgt. Um eine Zelle zu finden, in der diese vier spezifischen Aminosäuren gleichzeitig vorhanden sind, wären 1028 Bakterien nötig. Das ist die geschätzte Menge an Bakterien auf der Erde. Wie schafften es die Forscher, die Aktivität überhaupt zu finden, wenn die Wahrscheinlichkeit so klein ist? Der Grund dafür liegt darin, dass sie für ihre willkürliche Suche einen sehr kleinen Bereich (von 21 Aminosäuren) in einem richtig gefalteten Protein aus 460 Aminosäuren auswählten. Wenn alle 9 Aminosäuren (die nachgewiesenermaßen für die Aktivität verantwortlich sind) für die Aktivität wichtig wären, würde die Wahrscheinlichkeit, diese Aktivität zu finden, 2x10-12 betragen. Das bedeutet, dass statistisch zwei solcher Mutanten in der von ihnen erzeugten Population zu finden wären. Wenn nur die vier in allen aktiven Sequenzen nachgewiesenen Aminosäuren genügen würden, um die Aktivität zu verursachen, dann beträgt die Wahrscheinlichkeit 6x10-6. Dies bedeutet, dass ca. eine Million aktive Strukturen in der Population von Mutanten vorhanden sind.
|
|
Fazit
Was kann man aus diesen Studien lernen, wenn man die berühmte Nadel im Heuhaufen sucht? Liefern sie den Beweis, dass Zufallsprozesse funktionsfähige Proteine wie Enzyme mit der entsprechenden 3-dimensionalen Struktur erzeugen? Diese Untersuchungen sind interessant, aufschlussreich und sie unterstützen eigentlich die Schlussfolgerungen von Designkonzepten: Nach über 20 Jahren intelligenter Forschungsarbeit, der Anwendung moderner Methoden der Gentechnologie und dem Einsatz von Computermodellierung wurde, ausgehend von einem Enzym mit seiner natürlich gefalteten Struktur, mit eingeschränkten zufälligen Sequenzvariationen in einem kleinen Bereich des Proteins durch gelenkte Evolution eine Aktivität erzielt, die im Vergleich zur natürlichen mehrstufigen Reaktion 10 Millionen mal langsamer war. „Es ist keine starke Übertreibung, diesen experimentellen Aufwand damit zu vergleichen, dass man einen mit Sand gefüllten Trichter genauso über einen Strand bewegt, dass die Botschaft zu lesen ist: Hier ist der Beweis für Evolution. Zugegebenermaßen wäre es hierbei nicht nötig, dass für die Botschaft die exakte Lage jedes Sandkörnchens genau bestimmt wäre, aber die Ursache für die gesamte Erscheinung ist eindeutig nicht zufällig“ (Dieser Vergleich stammt von Truman, persönliche Mitteilung).
|
| Nach Tawfik ist die Entstehung funktioneller Proteine „etwas, das einem Wunder sehr ähnlich ist“. |
|
|
Dan Tawfik (2013) formuliert folgendermaßen: Wenn man ein Enzym identifiziert hat, das für eine gesuchte Reaktion eine schwache, unklare Aktivität aufweist, so ist ziemlich klar, dass man durch Mutationen und entsprechende Selektion diese Aktivität um mehrere Größenordnungen verbessern kann. Was fehlt, ist eine Hypothese für die früheren Etappen, in denen noch gar keine enzymatischen Aktivitäten vorliegen, also ohne aktive Zentren und Faltungen, bei denen Selektionsmechanismen als Ausgangspunkte ansetzen können. Evolution steckt in der Zwickmühle: Nichts kann sich entwickeln, bevor es nicht bereits besteht und funktioniert. Nach Tawfik ist die Entstehung funktioneller Proteine „etwas, das einem Wunder sehr ähnlich ist“. Ich würde sogar noch weiter gehen und sagen: Es ist ein Wunder.
|
Literatur
- Axe DD (2004)
- Estimating the prevalence of protein sequences adopting functional enzyme folds. J. Mol. Biol. 341, 1295-1315. doi:10.1016/jmb.2004.06.058.
- Axe DD (2010)
- The case against a Darwinian origin of protein folds. BIO-Complexity 2010(1):1-12. doi:10.5048/BIO-C.2010.1.
- Bartel DP & Szostak JW (1993)
- Isolation of new ribozymes from a large pool of random sequences. Science 261, 1411-1418. doi:10.1126/science.7690155.
- Chao F-A et al. (2013)
- Structure and dynamics of a primordial catalytic fold generated by in vitro evolution. Nat. Chem. Biol. 9, 81-83. doi:10.1038/nchembio.1138.
- Dessailly BH et al. (2009)
- PSI-2: Structural genomics to cover protein domain family space. Structure 17, 869-881. doi:10.1016/j.str.2009.03.015.
- Doi N, Kakukawa K, Oishi Y & Yanagawa H (2005)
- High solubility of random-sequence proteins consisting of five kinds of primitive amino acids. Prot. Eng. Des. Sel. 18, 279-284. doi:10.1093/protein/gzi034.
- Fox S & Kaoru W (1958)
- Thermal Copolymerization of Amino Acids to a Product Resembling Protein. Science New Series, Vol. 128, No. 3333 p. 1214.
- Gregory JS (2011)
- Kinetic Characterization of Single Strand Break Ligation in Duplex DNA by T4 DNA Ligase. J. Biol. Chem. 286, 44187-44196. doi: 10.1128/MCB.23.16.5919-5927.2003.
- Keefe AD & Szostak JW (2001)
- Functional proteins from a random-sequence library. Nature 410, 715-718. doi:10.1038/35070613.
- Kozulic B (2011)
- Proteins and Genes, Singletons and Species: http://vixra.org/abs/1105.0025.
- Leisola M & Turunen O (2007)
- Protein engineering: opportunities and challenges. Appl. Microbiol. Biotechnol. 75, 1225-1232. doi:10.1007/s00253-007-0964-2.
- Lo Surdo P, Walsh MA & Sollazo M (2004)
- A novel ADP- and zinc-binding fold from function-directed in vitro evolution. Nat. Struct. Mol. Biol. 11, 382-383. doi:10.1038/nsmb745.
- Reidhaar-Olson JF & Sauer RT (1990)
- Functionally acceptable substitutions in two α-helical regions of λ repressor. Proteins 7, 306-316. doi: 10.1002/prot.340070403.
- Riechmann L & Winter G (2000)
- Novel folded protein domains generated by combinatorial shuffling of polypeptide segments. Proc. Natl. Acad. Sci. 97, 10068-10073.
- Roberts RW & Szostak J (1997)
- RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. 94, 12297-12302.
- Seelig B & Szostak JW (2007)
- Selection and evolution of enzymes from a partially randomized non-catalytic scaffold. Nature, 448, 828-833. doi: 10.1038/nature06032.
- Silverman JA, Balakrishnan R & Harbury PB (2001)
- Reverse engineering the (β/α)8 barrel fold. Proc. Natl. Acad. Sci. 98, 3092-3097. doi:10.1073/pnas.041613598.
- Stomel JM et al. (2009)
- A man-made ATP binding protein evolved independent of nature causes abnormal growth in bacterial cells. PLoS ONE 4:e7385. doi:10.1371/journal.pone.0007385.
- Tawfik D (2013)
- http://www.asbmb.org/asbmbtoday/asbmbtoday_article.aspx?id=48961.
- Truman R (2013
- Information Theory - Part 4. Fundamental Theorems of Coded Information Systems Theory. J. Creation 27, 71-77. http://creation.com/cis-4.
- Yockey HP (1977)
- A calculation of the probability of spontaneous biogenesis by information theory. J. Theor. Biol. 67, 377-398. http://dx.doi.org/10.1016/0022-5193(77)90044-3.
-
|
|  |
